

Medulloblastoma

Originally named spongioblastoma cerebelli, the term “Medulloblastoma,” or cerebellar primitive neuro-ectodermal tumor, was first described by Cushing and Bailey in their seminal work classifying gliomas in 1924. Further characterization of this lesion occurred in 1983 when all malignant small cell tumors were classified as primitive neuroectodermal tumors (PNETs). The World Health Organization (WHO) continues to designate medulloblastoma as its own distinct entity, universally WHO grade IV.
Introduction
Medulloblastomas are the most common primary malignant brain tumor in the pediatric population, representing 20-40% of all pediatric brain tumors. They are uncommon in adults, most frequently observed in the third and fourth decade of life. The epidemiology of medulloblastoma varies dependent on subtype, with a peak incidence in infants and young children, followed by another peak in adults older than 16 years of age. This bimodal age distribution distinguishes the medulloblastoma subtype observed in infants/children from that of adults.
Although medulloblastoma is primarily a sporadic lesion, several familial cancer syndromes have been associated with this tumor, including Gorlin syndrome, Rubinstein-Taybi syndrome, Turcot syndrome, and Li-Fraumeni syndrome. There have also been multiple germline genetic mutations and signaling pathways implicated in the development and pathogenesis of medulloblastoma. These mutations include a disruption of the sonic hedgehog (Shh) or Wingless (Wnt) pathways, amplification of the Myc gene, loss of TP53 or ARF, and isochromosome 17q. Rare familial medulloblastomas have also been reported, involving mutation of human suppressor of fused homolog (SUFU gene).
The classification of medulloblastoma is composed of two characterizing schemes, molecular and histological. The combination of the histological and molecular group for each tumor has therapeutic implications as well as implications in patient outcomes.
The identification of specific transcriptomic differences in these lesions resulted in the classification of four medulloblastoma molecular subtypes with different prognostic implications. These molecular subtypes are as follows:
- Shh
- Wnt
- Group 3
- Group 4
The Wnt subtype has the most favorable prognosis, with a 5-year overall survival (OS) of 95%. The Shh and group 4 subtypes have a less favorable prognosis, 5-year OS of 75%, and the group 3 subtype has the worst prognosis at a 5-year OS of 50%. Mutations of TP53 are used to further stratify the Shh subtype with prognostic significance. Group 3 and group 4 medulloblastomas are not separated by a specific molecular etiology but by their histology.
Histologically, medulloblastoma tissue demonstrates round basophilic nuclei with prominent nucleoli and scant cytoplasm. Therefore, these tumors are categorized in the group of “small blue cell” tumors. Parenchymal invasion is commonly noted despite the presence of a pseudocapsule during dissection of the lesion.
Four histologic variants have been described by the WHO:
- Classic
- Desmoplastic/nodular (D/N)
- Large-cell/anaplastic (LCA)
- Medulloblastoma with extensive nodularity (MBEN)
These histologic variants can be used to classify tumors into two groups: desmoplastic tumors made up of D/N and MBEN variants, and LCA tumors. The LCA tumors are classified as group 3 and are associated with a worse clinical outcome relative to desmoplastic tumors. Generally, the classic histologic variant is more common in all molecular subtypes with less frequent presence of the other histological variants (Table 1).
| Subtype | Histology | Prognosis |
| WNT Subtype | Classic | Low-risk tumor |
| Large cell / anaplastic (rare) | Uncertain | |
| SHH Subtype, TP53-mutant | Classic | High-risk |
| Large cell / anaplastic | High-risk | |
| Desmoplastic / nodular (rare) | Uncertain | |
| SHH Subtype, TP53-wildtype | Classic | Standard-risk |
| Large cell / anaplastic | Uncertain | |
| Desmoplastic / nodular | Low-risk in infants | |
| Extensive nodularity | Low-risk in infants | |
| Non-WNT/non-SHH, Group 3 | Classic | Standard-risk |
| Large cell / anaplastic | High-risk | |
| Non-WNT/non-SHH, Group 4 | Classic | Standard-risk |
| Large cell / anaplastic (rare) | Uncertain |
Grossly, medulloblastomas commonly arise from the inferior medullary velum with cerebellar parenchymal or brainstem invasion. Cerebellar hemispheric involvement is more commonly observed in adults. Subarachnoid metastatic deposits, commonly referred to as “drop mets,” are often observed on the neural axis imaging. Of note, 10-35% of medulloblastomas demonstrate metastatic spread at the time of diagnosis. Much less frequently, approximately 5% of the patients demonstrate extraneural metastasis, such as bone marrow or peritoneal metastasis related to ventriculoperitoneal shunting.
Clinical Presentation
Medullablastomas often clinically manifest similar to other posterior fossa lesions, with headache, vomiting, lethargy, and gait instability. Cranial nerve palsies may occur secondary to mass effect or if the tumor has invaded the brainstem. Infants may present with macrocephaly, irritability, projectile vomiting, and upgaze palsy associated with hydrocephalus. Spinal metastatic lesions, while rare, may cause symptoms of cord compression, such as urinary retention, lower extremity weakness, and back pain.
Diagnosis
Diagnosis is initially made using imaging studies. Magnetic resonance (MR) imaging of the craniospinal axis with and without contrast allows the clinician to identify the extent of the tumor, its relation to adjacent structures, the encasement of vessels, and the invasion of cerebellar tissue and brainstem. Contrasted MR imaging demonstrates a heterogeneously enhancing and T1/T2 hypointense lesion.
Neural axis imaging to identify metastatic deposits is essential in the treatment of this disease as it appears to be the strongest predictor of poor outcome. MR spectroscopy can also be used to differentiate medulloblastoma from other cerebellar tumors. For additional discussion of radiographic characteristics of medulloblastoma, please refer to Medulloblastoma in the Neuroradiology sub-volume.
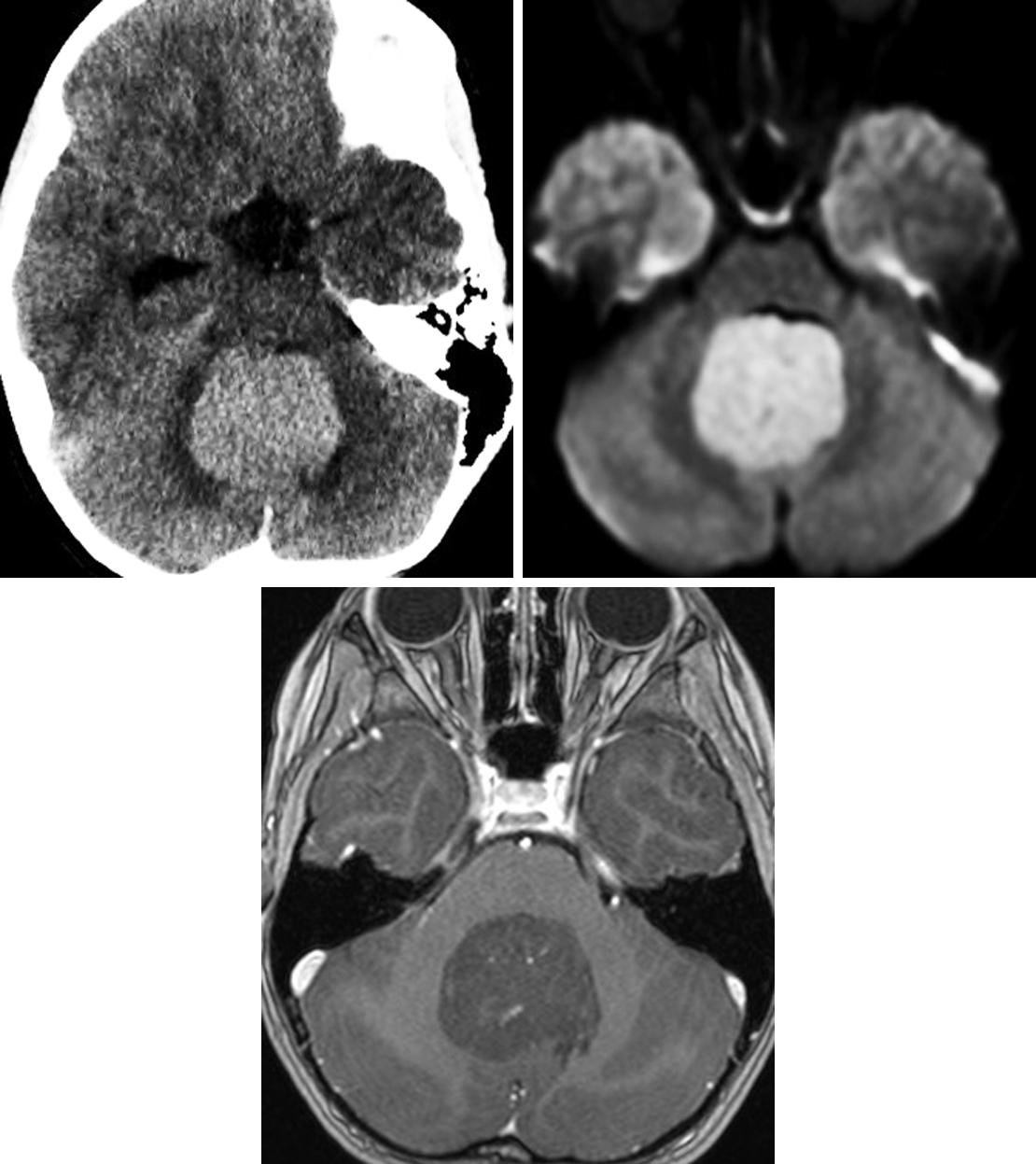
Figure 1: The hyperdensity of this medulloblastoma on CT (top row left) is typical and representative of the tumor's hypercellularity. Likewise, the DWI (top row right) shows restricted diffusion, also representing hypercellularity. This patient's lack of significant enhancement on this T1WI postcontrast image (bottom) is most typical of group 4 medulloblastomas. The intraventricular location is also typical, with these lesions sometimes visibly arising from the 4th ventricular roof.
In addition to imaging studies, a lumbar puncture with cytologic analysis of the cerebrospinal fluid (CSF) is completed either preoperatively or, in select cases, at least 10 days postoperatively. The presence of neoplastic cells in the CSF is indicative of metastases and is necessary for staging of the disease. Histopathologic analysis of the tumor sample is also used for staging.
Indications for Surgery
Surgical resection is indicated for medulloblastoma to relieve the associated mass effect, obtain tissue for pathological diagnosis, and reduce the tumor burden. This resection must be as extensive as a complete gross total resection but follows the principle of “maximally safe” resection. Use of neo-adjuvant chemotherapy in the setting of medulloblastoma to reduce the tumor size preoperatively is reported in the literature but has not been systematically reviewed in the pediatric population.
Preoperative Considerations
Initially, the majority of patients need to be treated for hydrocephalus. Temporary CSF diversion using an external ventriculostomy drain (EVD) provides relief from symptomatic hydrocephalus; drainage of CSF intraoperatively permits improved posterior fossa decompression and visualization. The EVD placement is not necessary in all patients as preoperative steroid administration may be sufficient to alleviate symptoms via a reduction in inflammation and vasogenic edema.
EVDs are needed primarily in young children and those with extensive disease and are rarely used in children older than 10 years of age. Permanent shunts are not recommended as most resections can preclude the need for a permanent CSF diversion postoperatively. Ventriculoperitoneal shunts can also result in peritoneal seeding with neoplastic cells.
The precise location of the tumor and its relation to the vital structures of the posterior fossa should be characterized on preoperative imaging. Use of intraoperative neuronavigation may be useful in navigating the posterior fossa contents safely during the dissection. Attention should be paid to the tumor’s relation to the brainstem, fourth ventricle, deep nuclei, and posterior inferior cerebellar artery (PICA). Electrophysiologic monitoring may be used in select tumors to minimize the manipulation of critical neural structures.
RESECTION OF MEDULLOBLASTOMA
The goal of surgery is to obtain a tissue diagnosis and to perform a maximal safe resection. Patients who suffer from medulloblastoma are classified as average-risk or high-risk based on age at diagnosis, evidence of metastasis, and residual tumor volume. Near total or less than 1.5-cm2 residual tumor within the image slice with maximal residual tumor allows the patient to be upgraded to average risk (Staging system proposed by Packer and colleagues) and has a significant impact on overall 5-year progression-free survival. Importantly, the surgeon should not justify significant neurological injury to achieve the 1.5-cm2 threshold.
The surgical approach to these lesions can involve a standard midline suboccipital approach, telovelar, transvermian, or lateral transcortical transcerebellar approach. During operative positioning of the children greater than 4 years of age, skull clamps with pediatric pins may be used safely; however, in younger children, a padded horseshoe with eye protection is preferred.
Patient positioning is variable and dependent on the surgeon’s preference and the choice of approach. Sitting, prone, and lateral positions have been described. The lateral or park-bench positions are often preferred due to the lower risk for associated morbidity.
A sitting position is associated with a higher risk of air embolism as well as subdural and operative bed hematomas. Nonetheless, using this position, favorable viewing angles have been described for tumors within the posterior third ventricle, quadrigeminal cisterns, pineal region, superior cerebellar surface, and tentorium.
Based on the location of the tumor mass, I most often prefer a standard midline suboccipital craniotomy. Wide draping should incorporate the Frazier’s point in the event of intraoperative swelling that would require an emergent temporary CSF diversion using an external ventricular drain.
In children, the surgeon must be mindful of the presence of large midline dural venous lacunae or sinus (occipital sinus) or other venous lakes adjacent to the other venous sinuses as these structures can lead to significant life-threatening hemorrhage if damaged or entered inadvertently with the drill.
Rarely, a C1 hemilaminectomy may be needed to allow visualization of the obex if the tonsils are pushed inferiorly. During this hemilaminectomy, one must be careful not to inadvertently open the dura as the C1 lamina is cartilaginous and incompletely fused in the midline among children.
If the tumor is located in the lateral cerebellar hemispheres or pontocerebellar angle, a lateral suboccipital craniotomy may be required. For this approach, I plan the vertical incision between the midline and mastoid process, with the exact location dependent on the location of the tumor. The bone flap is situated just below the level of the transverse sinus and is smaller and more circular. This flap is elevated using a single burr on the inferior edge of the transverse sinus and medial to the transverse-sigmoid junction.
The dura is opened in the standard “Y” fashion. The paramedian incisions can be extended superolaterally toward the transverse-sigmoid junction as needed, and the midline incision should extend inferiorly to the lower limit of the tonsils. The dural flaps are then reflected and tacked up by means of nylon sutures.
The tumor is commonly identified in the midline and often extends through the foramen of Magendie. It is important to identify the pseudo-margins of the tumor in all planes; these margins are often challenging to maintain during dissection.
The surgeon should be cognizant that maximal safe resection is the goal; the tumor should not be followed into the brainstem. Therefore, it is imperative to identify the brainstem-tumor interface, after which the lateral and rostral planes are also delineated. If identification of these planes is difficult, extra care should be used to avoid entering the deep nuclei, fourth ventricle, or cerebellar peduncles. After identification of these planes, the feeding vessels should be coagulated prior to resection to minimize bleeding.
Circumferential dissection of the tumor is then conducted, followed by central debulking if necessary, preserving the en passage vessels perfusing eloquent parenchyma. An ultrasonic aspirator (CUSA) is often useful during this step. Resection can be guided by neuronavigation, particularly in determining the depth of resection, but the surgeon must be mindful of brain sag and its impact on the accuracy of neuronavigation.
Closure
Once maximally safe resection is complete, hemostasis is obtained. Judicious use of bipolar electrocautery is recommended. Retention of hemostatic agents is not recommended so as to prevent enhancing artifact on postoperative MR imaging that can resemble recurrent/residual tumor. The dura is closed in a watertight fashion via an autologous or synthetic dural graft.
Postoperative Considerations
During the immediate postoperative period, dysmetria, ataxia, and rarely lower cranial nerve paresis may be transiently present; these commonly resolve within days to weeks. A major complication of posterior fossa surgery that can be observed following resection of a medulloblastoma via the transvermian route is cerebellar mutism, also referred to as posterior fossa syndrome.
Cerebellar mutism is theorized to result from injury to the dentatorubrothalamic tract. This entity is characterized by delayed-onset oropharyngeal apraxia and can also involve cranial nerve deficits. It is commonly observed after surgery among up to 30% of the patients that undergo resection. This syndrome can resolve within the acute postoperative period or lead to permanent neurocognitive deficits.
Acute hydrocephalus can present during the postoperative period. An EVD will be necessary, permitting a weaning trial evaluating the need for permanent diversion of CSF.
Pain must be adequately controlled in children during the postoperative period to avoid anxiety and hyperventilation, which can lead to an increase in intracranial pressure.
Adjuvant Treatment
Postoperative radiation therapy should encompass the entire neural axis, including focal treatment of the resection cavity and any of the spinal metastases. The use of radiation in children is associated with undesirable consequences such as neuropsychological impairment and endocrinopathies. Therefore, radiation is very carefully considered in children younger than 3 years of age. Radiation therapy is most effective if completed soon after surgery, and a delay beyond 28 days negatively impacts the patient’s prognosis.
Chemotherapy alone is the only adjuvant treatment for children younger than 3 years of age. These patients receive a myeloablative chemotherapy regimen with stem cell rescue only. In those receiving radiation, chemotherapy has permitted a reduction in radiation dosage without affecting overall outcomes. This is especially beneficial in that radiation is responsible for the majority of long-term sequelae associated with medulloblastoma.
Several prognostic factors have been identified for determination of clinical outcomes. These include age (whether the patient is younger than or older than 3 years), postoperative residual tumor volume, disseminated neuroaxis disease, and neoplastic cells on spinal fluid cytology.
After resection of the tumor, the patients are staged as either average risk or high risk, based on MR imaging and CSF cytology results, and age. Average risk is defined as gross total resection less than 1.5 cm2 residual in a single image of a cross sectional imaging series and no evidence of metastatic disease. High risk is defined as significant residual tumor greater than 1.5 cm2 residual in a single image of a cross sectional imaging series or presence of metastatic lesions. Average risk conveys a 5-year survival rate of greater than 5% and progression-free survival rate of 50%. High risk conveys a poor prognosis with a 5-year progression-free survival of 35-50%.
This classification will determine the radiation therapy and chemotherapy courses. These regimens have improved the 5-year overall and event-free survival of both groups to greater than 80% and have resulted in a cure rate of 70-75% in children 3 years of age or older. Despite these improvements, the patients who survive will likely experience long-term sequelae and a reduced quality of life.
Pearls and Pitfalls
- Maximal safe resection is the key in management of medulloblastoma, significantly impacting the 5-year survival rate. The brainstem-tumor interface should be respected, and caution should be taken to prevent severe cranial nerve deficits and associated co-morbidities.
- Preoperative MRI scans of the neural axis should be acquired to prevent artifactual diagnosis of drop-metastasis from operative debris.
- Persistent occipital sinus is present in 10% of adults and frequently in infants. Caution must be exercised when incising the dura to avoid life-threatening bleeding; the sinus may require ligation.
Contributors: Adam Leibold BS, Benjamin K. Hendricks MD, Farhan A. Mirza MBBS
References
Albright AL, Adelson PD, Pollack IF (Eds). Medulloblastomas. In: Principles and Practice of Pediatric Neurosurgery. 3rd ed. New York: Thieme, 2008.
Quinones-Hinojosa A, Raza SM, Laws ER (Eds). Controversies in Neuro-Oncology: Best Evidence Medicine for Brain Tumor Surgery. 1st ed. New York: Thieme, 2013.
Gajjar AJ, Robinson GW. Medulloblastoma—translating discoveries from the bench to the bedside. Nature Reviews Clinical Oncology Nat Rev Clin Oncol. 2014;11(12):714-722.
Kunschner LJ. Harvey Cushing and medulloblastoma. Arch Neurol. 2002;59(4):642-5.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (Eds). WHO Classification of Tumours of the Central Nervous System (Revised 4th Edition). Lyon: IARC, 2016.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2016) World Health Organization Histological Classification of Tumours of the Central Nervous System. Acta Neuropathologica. 2016;131(6):803–820
Ostling L, Raffel C. Medulloblastoma. Pediatric Neurosurgery: Tricks of the Trade. Alan Cohen (Ed.). New York: Thieme, 2016.
Please login to post a comment.




